in Candida Yeast Species
Table of Contents
Background
The GAL1, GAL7, and GAL10 genes encode the three enzymes of the Leloir pathway, the principal fungal route for converting galactose into glucose-1-phosphate for entry into glycolysis. In Saccharomyces cerevisiae, these genes are physically clustered on chromosome II and tightly co-regulated, an arrangement broadly conserved across the fungal kingdom. However, whether this genomic neighbourhood is maintained in more distantly related Candida species has remained poorly understood.

Research Goal
This project aimed to locate the three GAL genes within a draft genome of an unknown Candida species, compared protein-level conservation across taxa, and analysed the surrounding chromosomal architecture.
Analysis:
Genomic Locations
Gene coordinates were identified within two well characterised Candida species using NCBI Genome Annotation pages and confirmed with assembly-specific filtering.
| Gene | C. albicans (Chr 1) | S. cerevisiae (Chr 2) |
|---|---|---|
| GAL1 | NC_032089.1:449253–450800 | NC_001134.8:279021–280607 |
| GAL10 | NC_032089.1:452164–454191 | NC_001134.8:276253–278352 |
| GAL7 | NC_032089.1:456751–457911 | NC_001134.8:274427–275527 |
All three genes co-localize to a single chromosome in both species, consistent with their role as a co-regulated cluster.
Protein Sequence Similarity (C. albicans vs. S. cerevisiae)
Pairwise alignments were performed with the UniProt Align Tool (Clustal Omega) and verified with EMBOSS Needle (BLOSUM62).
| Gene | Similarity | Identity | Notes |
|---|---|---|---|
| GAL7 | ~75% | ~60% | Highest conservation |
| GAL10 | ~71% | ~53.6% | |
| GAL1 | ~62% | ~44.9% | Lowest conservation |
GAL7 was selected for a broader cross-Candida BLAST analysis. A UniProt BLAST search against Candida species returned 27 results with 13 highly significant hits (E-value = 0.0). Multiple-sequence alignment of these hits showed protein identity across all species is ~81.7%, indicating strong conservation of GAL7 function across the genus.
| XP_713769.2 | B9W705_CANDC | M3IKC2_CANMX | C5MEX8_CANTT | A0A8H7ZK36 | A0AAD5BIS8 | A0A9W4XB10 | H8X1P2_CANO9 | A0AAI9SXI8 | G8B7Z7_CANPC | A0A367XTK6 | A0A367Y509 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| XP_713769.2 | 100.00% | 96.37% | 82.69% | 84.60% | 80.00% | 79.18% | 79.74% | 79.11% | 79.11% | 78.90% | 82.19% | 82.19% |
| B9W705_CANDC | 96.37% | 100.00% | 83.52% | 84.33% | 80.00% | 79.18% | 79.74% | 79.37% | 78.85% | 78.63% | 82.47% | 82.47% |
| M3IKC2_CANMX | 82.69% | 83.52% | 100.00% | 82.14% | 79.01% | 79.34% | 77.01% | 78.85% | 77.96% | 78.51% | 83.52% | 83.24% |
| C5MEX8_CANTT | 84.60% | 84.33% | 82.14% | 100.00% | 78.42% | 76.92% | 74.93% | 77.02% | 76.96% | 75.55% | 88.52% | 88.25% |
| A0A8H7ZK36 | 80.00% | 80.00% | 79.01% | 78.42% | 100.00% | 93.37% | 79.63% | 95.00% | 80.74% | 91.41% | 77.35% | 77.90% |
| A0AAD5BIS8 | 79.18% | 79.18% | 79.34% | 76.92% | 93.37% | 100.00% | 79.50% | 95.33% | 80.49% | 92.03% | 76.37% | 76.92% |
| A0A9W4XB10 | 79.74% | 79.74% | 77.01% | 74.93% | 79.63% | 79.50% | 100.00% | 79.42% | 77.84% | 79.78% | 74.52% | 74.79% |
| H8X1P2_CANO9 | 79.11% | 79.37% | 78.85% | 77.02% | 95.00% | 95.33% | 79.42% | 100.00% | 79.84% | 92.86% | 76.71% | 77.26% |
| A0AAI9SXI8 | 79.11% | 78.85% | 77.96% | 76.96% | 80.74% | 80.49% | 77.84% | 79.84% | 100.00% | 81.37% | 75.27% | 75.82% |
| G8B7Z7_CANPC | 78.90% | 78.63% | 78.51% | 75.55% | 91.41% | 92.03% | 79.78% | 92.86% | 81.37% | 100.00% | 76.65% | 76.92% |
| A0A367XTK6 | 82.19% | 82.47% | 83.52% | 88.52% | 77.35% | 76.37% | 74.52% | 76.71% | 75.27% | 76.65% | 100.00% | 97.54% |
| A0A367Y509 | 82.19% | 82.47% | 83.24% | 88.25% | 77.90% | 76.92% | 74.79% | 77.26% | 75.82% | 76.92% | 97.54% | 100.00% |
PIPELINE
-ax splice preset aligned the mRNA sequences to the draft genome. All three genes were mapped to "scaffold_1".minimap2-ax splice
#!/bin/bash
module load minimap2
minimap2 -ax splice Unknown-Yeast-Genome.fasta GAL_mRNA.fasta > GAL_alignment.sam
samtools viewsamtools sortsamtools index
#!/bin/bash
module load samtools
samtools view -O BAM -o gal_alignments.bam GAL_alignments.sam
samtools sort -O BAM -o gal_alignments.sort.bam gal_alignments.bam
samtools index -b gal_alignments.sort.bam
samtools view gal_alignments.sort.bam | cut -f1,2,3,4samtools faidx then aligned with nucmer and visualized with mummerplot.nucmermummerplot
#!/bin/bash
module load samtools
module load anaconda3/2023.09
source activate mummer4
samtools faidx cAlb_genome.fna "NC_032089.1" > CAlb_Chr1.fasta
samtools faidx Unknown-Yeast-Genome.fasta "scaffold_1" > UnknownYeast_scaffold1.fasta
nucmer -t 2 --mum -p chr1_vs_scaffold1 CAlb_Chr1.fasta UnknownYeast_scaffold1.fasta
mummerplot -t png --filter -R CAlb_Chr1.fasta -Q UnknownYeast_scaffold1.fasta \
-p chr1_vs_scaffold1_plot chr1_vs_scaffold1.deltaMUMmer4pythoncirca 2.0
#!/bin/bash
module load anaconda3/2023.09
source activate mummer4
nucmer -t 4 --mum -p mum_alignment cAlb_genome.fna Unknown-Yeast-Genome.fasta
mummerplot -t png --filter -Q Unknown-Yeast-Genome.fasta -R cAlb_genome.fna \
-p genome_plot mum_alignment.delta #!/usr/bin/env python3
import csv
with open("out.coords") as infile, open("circa_links.csv", "w", newline="") as outfile:
# skip 4 header lines
for _ in range(4):
infile.readline()
writer = csv.writer(outfile)
writer.writerow(["chromosome1", "start1", "end1", "chromosome2", "start2", "end2"])
for line in infile:
fields = line.strip().split("\t")
if len(fields) < 9:
continue
s1, e1, s2, e2 = fields[0], fields[1], fields[2], fields[3]
chr1, chr2 = fields[-2], fields[-1]
writer.writerow([chr1, s1, e1, chr2, s2, e2]) #!/bin/bash
# Combine both into one file
awk '{print $1","$2}' cAlb_genome.fna.fai > chromosome_lengths.csv
awk '{print $1","$2}' Unknown-Yeast-Genome.fasta.fai >> chromosome_lengths.csv
# Add header
sed -i '1s/^/chromosome,length\n/' chromosome_lengths.csvResults:
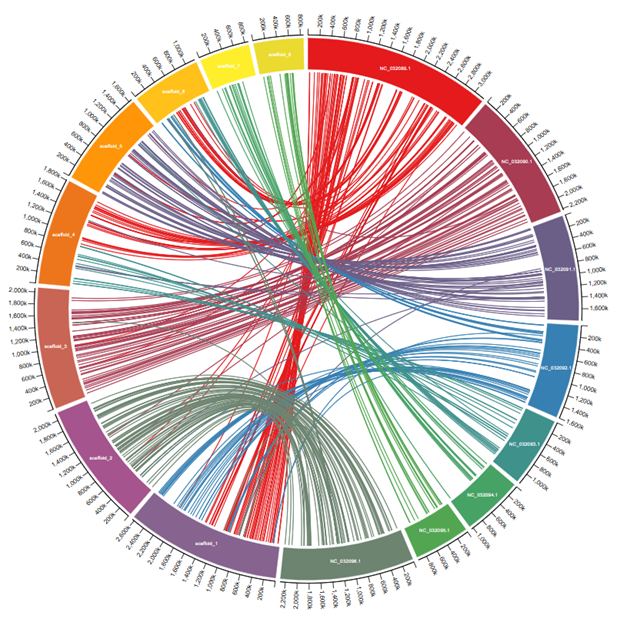
Mapping to the unknown Candida draft genome confirmed that the GAL cluster order (GAL1 → GAL10 → GAL7) is preserved on scaffold_1, although the cluster is located on an inverted region relative to C. albicans.

To verify the gene mapping, the Minimap2 alignment output was viewed with SAMtools again, but with the flag character included to determine strand direction [5,6]. GAL1 (XM_708671.2) showed alignment on the reverse strand (FLAG = 16), indicating there may be a different transcriptional orientation [6,7]. Overall the GAL gene cluster order is still conserved, but may be within or outside a larger inversion of the chromosome region relative to the reference chromosome
| Gene | mRNA ID | Scaffold | Start pos. | Strand |
|---|---|---|---|---|
| GAL1 | XM_708671.2 | scaffold_1 | 476,277 | Reverse (FLAG=16) |
| GAL10 | XM_708673.2 | scaffold_1 | 478,496 | Forward |
| GAL7 | XM_708676.2 | scaffold_1 | 483,668 | Forward |
At the whole-genome level, scaffolds 2 and 4 show complete inversions relative to C. albicans chromosomes 2 and 8, and multiple scaffolds align to chromosome 1: evidence of translocations or chromosomal fragmentation.

The GAL cluster protein sequence is highly conserved (~ 80% identity across Candida), while the surrounding genomic architecture has diverged substantially. This contrast highlights how functional constraint at the sequence level can persist even as chromosomal structure undergoes significant reorganization.

Key Findings
By combining sequence homology searches, synteny analysis, and multi-species protein alignments, the study shows that while the Gal1, Gal7, and Gal10 proteins are highly conserved at the amino acid level, their chromosomal context has undergone considerable reorganisation relative to S. albicans. This contrast between gene-level conservation and large-scale genomic rearrangement highlights the value of integrating both levels of analysis when characterising metabolic pathways in non-model organisms.
Bioinformatics Tools:
- NCBI Genome Annotation, UniProt BLAST & Align, EMBOSS Needle
- Minimap2 (splice-aware alignment), SAMtools, MUMmer4 / nucmer, mummerplot
- Circa 2.0 (Circos synteny plots)
- Python scripting (coordinate parsing, CSV formatting)
- HPC / SLURM job scheduling (Centaurus cluster)
- BAM/SAM processing, dot-plot interpretation
References:
- Altschul et al. (1990). Basic local alignment search tool. J. Mol. Biol., 215(3), 403–410.
- Krzywinski et al. (2009). Circos: An information aesthetic for comparative genomics. Genome Research, 19(9), 1639–1645.
- Kurtz et al. (2004). Versatile and open software for comparing large genomes. Genome Biology, 5(2), R12.
- Li, H. (2018). Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics, 34(18), 3094–3100.
- Li et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16), 2078–2079.