Table of Contents
Background
MicroRNAs in Gene Regulation
MicroRNAs (miRNAs) are small, non-coding RNA molecules (~18–25 nucleotides) that regulate gene expression post-transcriptionally by binding to complementary sequences in the 3’untranslated regions (3’UTRs) of target mRNAs. This binding recruits the RNA-Induced Silencing Complex (RISC), which either cleaves the target mRNA or represses its translation, depending on the degree of complementarity and the specific RISC proteins involved.
 BioRender.")
In animals, miRNA regulation is typically initiated with a cleavage event in the nucleus: long primary transcripts (pri-miRNA) are processed by the enzyme Drosha to produce ~70-nucleotide precursor hairpins (pre-miRNA). These precursors are exported to the cytoplasm, where the enzyme Dicer cleaves them into the mature, double-stranded miRNA duplex. One strand is selected (guide strand) and loaded into RISC, where it directs the complex to complementary targets. The mature miRNA typically exhibits a conserved seed region (nucleotides 2–8) that is essential for target recognition.
Bioinformatic Challenges in miRNA Research
Identifying miRNA targets computationally poses several challenges:
- Sequence degeneracy: Animal miRNAs often tolerate mismatches outside the seed region, meaning exact complementarity is not required. This results in high false-positive rates when using simple sequence matching.
- Structural conservation vs. sequence divergence: miRNA precursors are conserved primarily in their secondary structure (the hairpin stem) rather than their primary sequence. Homologous miRNAs from different species may share little overall sequence identity but preserve critical base-pairing interactions.
- Identifying the mature miRNA: The mature miRNA sequence is excised from the pre-miRNA hairpin, but the exact boundaries are sometimes ambiguous without functional validation. Computational tools must infer where the mature product begins and ends based on conserved structural features.
- Biological vs. computational truth: A miRNA may be computationally predicted to target a gene, but functional validation (e.g., reporter assays, AGO-CLIP sequencing) is required to confirm the interaction.
The Seed Region as a Predictive Feature
The miRNA seed region (positions 2–8 of the 5’ end of the mature miRNA) is the most conserved and functionally critical portion of the molecule. Seed region complementarity is usually sufficient for target recognition in animal miRNAs, whereas plant miRNAs typically require near-perfect complementarity across the entire miRNA. By extracting the seed region and using it to construct a profile Hidden Markov Model (HMM), we can search for overrepresented target motifs in 3’UTRs while accounting for the inherent variability of sequence matches.

Research Questions:
Given an alignment of precursor miRNA sequences from a family of related miRNAs, can we:
- Infer the conserved secondary structure using covariance analysis?
- Identify and extract the mature miRNA seed region?
- Build an HMM profile that captures the signal of true targets?
- Screen the Drosophila 3’UTRome and identify putative target genes?
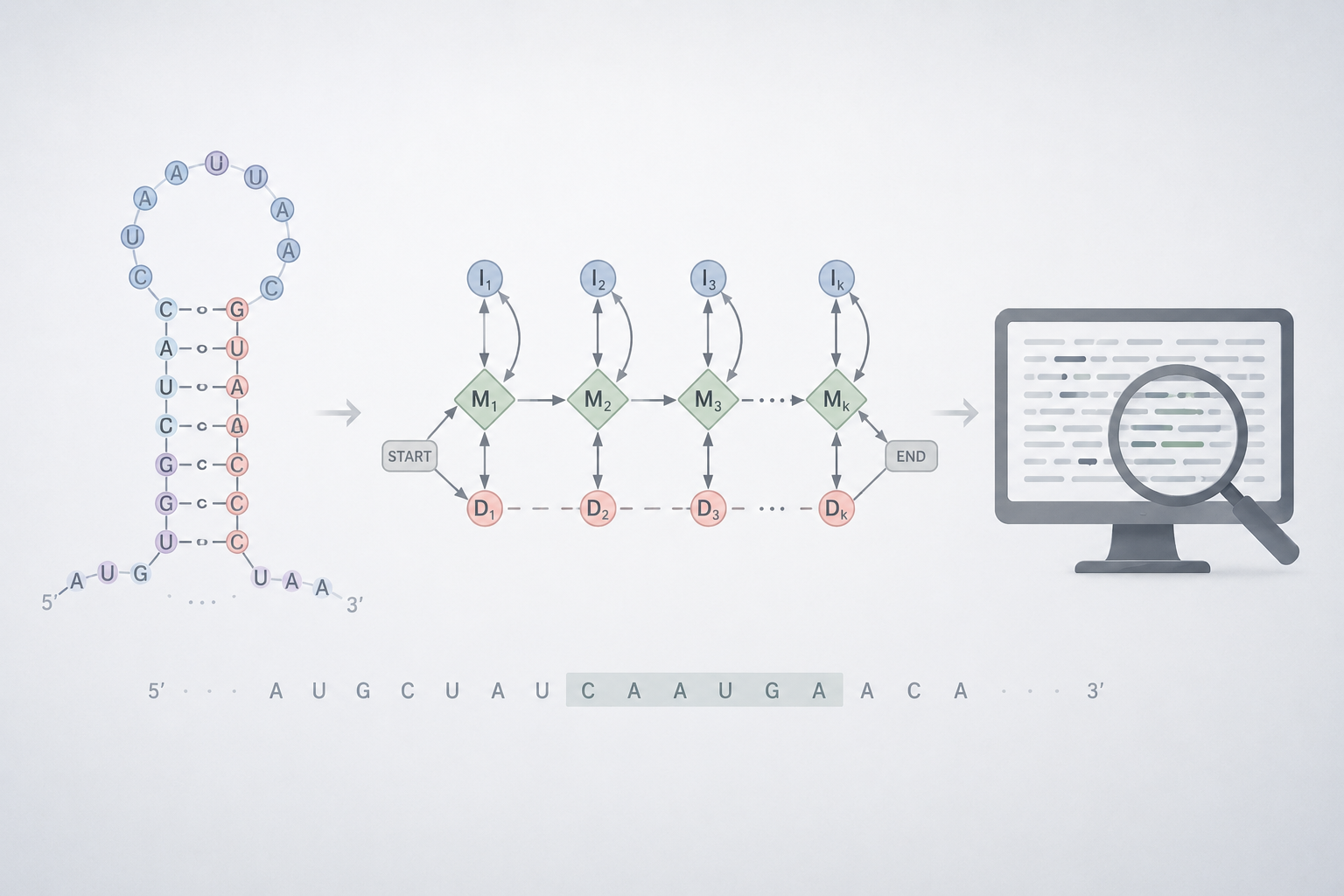
PIPELINE
R-scape
Python
#!/usr/bin/env python3
"""
Extract a column region from a Stockholm alignment file, reverse transcribe
(U to T), and optionally save the reverse complement to the output file.
"""
import argparse
COMPLEMENT = str.maketrans('ATGCatgc', 'TACGtacg')
parser = argparse.ArgumentParser(
description="Extract a column region from a Stockholm alignment, reverse transcribe, "
"and optionally reverse complement the sequences."
)
parser.add_argument(
"--stockholm_file",
default="data/RF00754.stockholm.txt",
help="Path to the input Stockholm alignment file"
)
parser.add_argument(
"--start", "-s",
type=int,
default=12,
metavar="COL",
help="First alignment column to extract, 1-indexed (default: 12)"
)
parser.add_argument(
"--end", "-e",
type=int,
default=32,
metavar="COL",
help="Last alignment column to extract, 1-indexed inclusive (default: 32)"
)
parser.add_argument(
"--rc",
action="store_true",
help="Save the reverse complement of each sequence to the output file"
)
parser.add_argument(
"--output", "-o",
default="data/mature_region.sto",
metavar="FILE",
help="Output Stockholm file name (default: mature_region.sto)"
)
def reverse_complement(seq):
"""Return the reverse complement of a DNA sequence, preserving gap characters."""
return seq.translate(COMPLEMENT)[::-1]
def parse_stockholm(stockholm_file):
"""Read a Stockholm file and return an ordered dict of {name: sequence}."""
sequences = {}
with open(stockholm_file) as f:
for line in f:
line = line.rstrip('\n')
if not line.strip() or line.startswith('#') or line.startswith('//'):
continue
parts = line.split()
if len(parts) == 2:
name, seq = parts
sequences[name] = seq
return sequences
def process_sequences(sequences, col_start, col_end, rc):
"""Extract columns, reverse transcribe, and optionally reverse complement."""
rt_label = "Rev. Complement" if rc else "Rev. Transcribed"
header = f"{'Sequence Name':<45} {'Extracted (cols {}-{})'.format(col_start, col_end):<25} {rt_label}"
print(header)
print("-" * len(header))
results = {}
for name, seq in sequences.items():
extracted = seq[col_start - 1 : col_end]
rt = extracted.replace('U', 'T').replace('u', 't')
final = reverse_complement(rt) if rc else rt
results[name] = final
print(f"{name:<45} {extracted:<25} {final}")
return results
def write_stockholm(results, output_file):
"""Write processed sequences to a Stockholm format file."""
with open(output_file, 'w') as f:
f.write("# STOCKHOLM 1.0\n")
for name, seq in results.items():
f.write(f"{name:<40} {seq}\n")
f.write("//\n")
print(f"\nSaved {len(results)} sequences to '{output_file}'")
def main():
args = parser.parse_args()
if args.start < 1:
parser.error("--start must be >= 1")
if args.end < args.start:
parser.error("--end must be >= --start")
sequences = parse_stockholm(args.stockholm_file)
results = process_sequences(sequences, args.start, args.end, args.rc)
write_stockholm(results, args.output)
if __name__ == "__main__":
main()
HMMER
#!/bin/bash
module load hmmer
hmmbuild results/miRNA.hmm data/mature_region.sto

nhmmer
#!/bin/bash
module load hmmer
nhmmer --tblout results/results.tbl results/miRNA.hmm data/FlyBase_GLCCGA.fasta
Results:
Covariance Structure: R-scape identified significantly covarying base pairs (positions 5–93, 6–92) confirming a conserved hairpin structure.


Seed Region: Positions ~5–25 on the 5’arm were selected as the mature miRNA candidate based on covariation support, alignment quality, and appropriate length (~21 nt).
Initial Search: Searching with positions from the 5’arm (U→T, no reverse complement) returned zero hits against Drosophila 3’UTRs.
Refined Search: After systematic adjustment, positions 64–83 from the 3’arm (reverse-complemented) yielded one hit:

| Target Gene | FBtr ID | E-value | Bit Score |
|---|---|---|---|
| Unknown | FBtr0075008 | 3.9–5.2 | ~12.6 |
The alignment shows partial complementarity consistent with miRNA targeting, but the high E-value indicates weak statistical confidence and possible chance alignment.
Key Findings & Biological Significance
A single putative miRNA target gene was identified (FBtr0075008) with marginal statistical support.
The low bit score and relatively high E-value indicate that this hit should be considered a preliminary prediction requiring functional validation. The weak signal may reflect:
- True biological targeting but with high sequence degeneracy
- A chance alignment that passes the threshold by coincidence
- An incorrect boundary for the miRNA seed region, despite careful structural analysis
Limitations: This analysis provides a preliminary prediction without functional validation. A more robust approach would use iterative searches with varying E-value thresholds, compare results against established databases (miRBase, TargetScan), and experimentally validate top candidates (reporter assays in Drosophila S2 cells).
Next steps: Validate FBtr0075008 against known miRNA target databases; extract and search seed-only regions (positions 2–8); integrate evolutionary conservation across Drosophila species to improve specificity.
Bioinformatics Tools:
| Tool | Purpose | Category |
|---|---|---|
| R-scape | Covariance-based secondary structure prediction for RNA | Structure Prediction |
| Python | Custom scripting (U→T conversion, reverse complementation) | Programming |
| HMMER | Hidden Markov Model construction and profile matching | Sequence Modeling |
| nhmmer | HMMER tool optimized for nucleotide-level searches | Sequence Search |
References:
- Bartel, D. P. (2009). MicroRNAs: Target Recognition and Regulatory Functions. Cell, 136(2), 215–233.
- Eddy, S. R. (2011). Accelerated Profile HMM Searches. PLoS Computational Biology, 7(10): e1002195.
- FARID, H. (2026). MicroRNA Biogenesis Pathway. https://app.biorender.com/profile/template/details/t-65cf2ea192cebb9ef433ce92-microrna-biogenesis-pathway
- Kozomara, A., Birgaoanu, M., & Griffiths-Jones, S. (2019). miRBase: from microRNA sequences to function. Nucleic Acids Research, 47(D1), D155–D162.